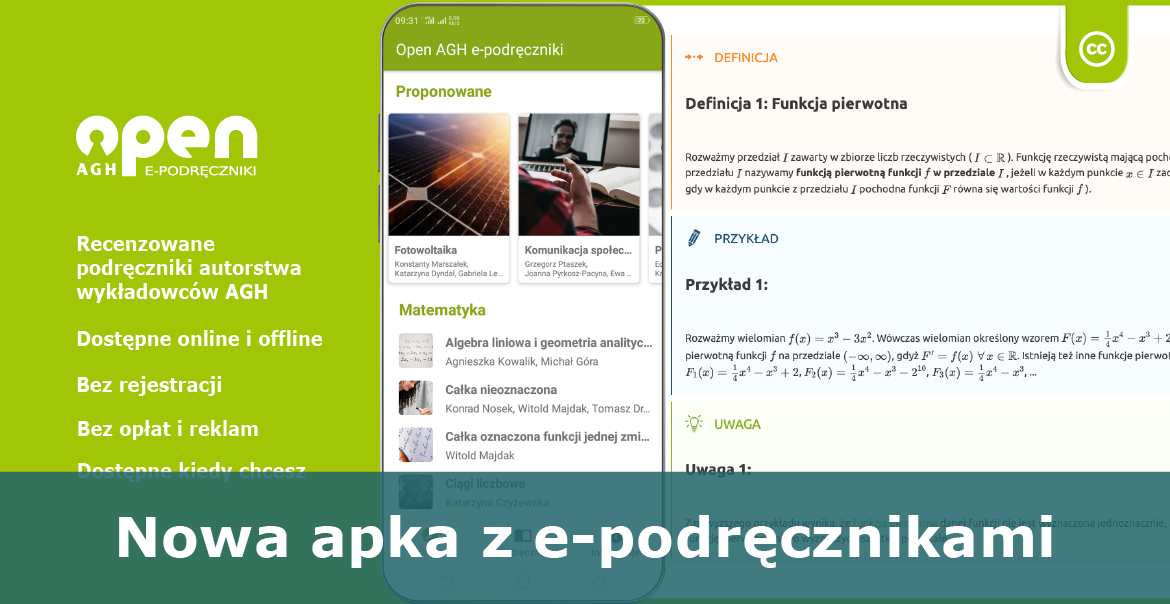

Najlepiej oceniane zasoby






Nasz serwis to ponad 1000 godzin nauki
Z naszych zasobów miesięcznie korzysta ponad 4500 osób
Dzięki naszym zasobom zdanych zostało 921547 egzaminów :-)

4 Zasoby

4 Zasoby

14 Zasobów

16 Zasobów

2 Zasoby

6 Zasobów

3 Zasoby

1 Zasób




